2 Preprocessing Ribosome footprints sequencing data
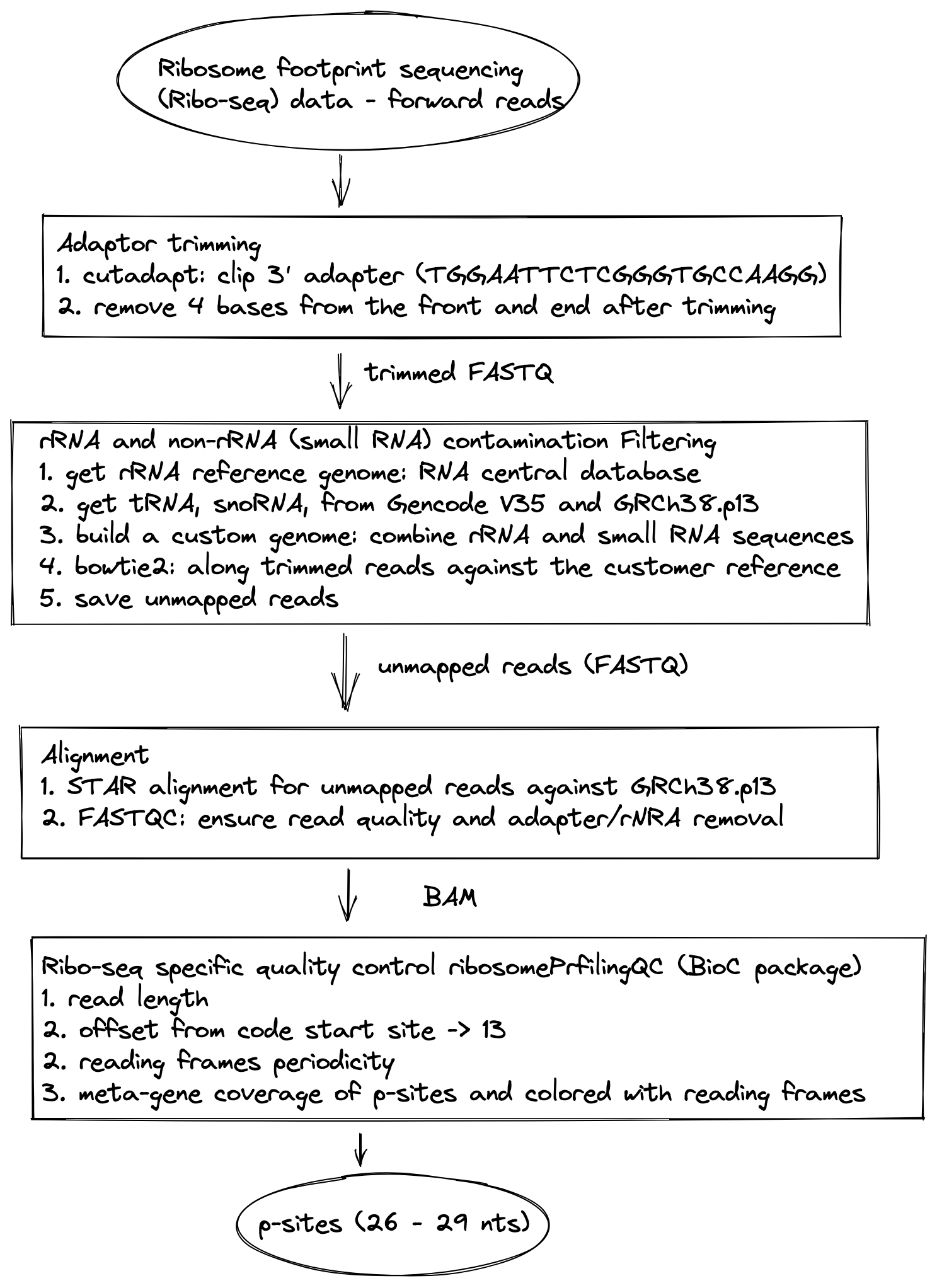
Figure 2.1: Flowchart of Ribo-seq data preprocessing
The script provided performs the preprocessing steps of our ribo-seq data. The following sections outline the preprocessing steps included in the shell script.
Software:
- FastQC/0.11.9-Java-11
- cutadapt/2.7-GCCcore-8.3.0-Python-3.7.4
- Bowtie2/2.4.1-GCCcore-8.3.0
- SAMtools/1.10-GCCcore-8.3.0
- STAR/2.7.6a-foss-2019b
2.1 Adapter trimming
Trimming forward 3’ adapter TGGAATTCTCGGGTGCCAAGG and removing 4 bases from the front and end after trimming:
# for forward read, trim 3' adapter TGGAATTCTCGGGTGCCAAGG
cutadapt --cores=4 -O 13 -a N{4}TGGAATTCTCGGGTGCCAAGG \
-u 4 -m 25 \
--untrimmed-output ${sample_name}_R1_untrimmed.fastq.gz \
-o ${sample_name}_R1_trimmed.fastq.gz \
$fq1 > ${sample_name}_cutadapt_log.txt
fq=$trim_dir/${sample_name}_R1_trimmed.fastq.gz The -O 13 is the minimum overlap for an adapter match, where the 13 is computed as 9 plus 4 (where 9 is the minimum overlap and 4 is the length of the unknown section). If you do not specify it, the adapter sequence would match the end of every read (because N matches anything), and ten bases would then be removed from every read.
This step yielded reads with 25 - 29 nt length.
2.2 Remove rRNA and non-rRNA contamination using Bowtie2
We first used RNA central database to identify rRNA sequences and built an rRNA reference genome rRNA_ref. Second, we used GFF of Gencode v35 to extract small RNA (tRNA, snoRNA, sRNA, microRNA, etc) sequences and customized a reference genome rsoRNA_ref. We then used bowtie2 to align the trimmed reads against the rRNA reference genome. Next, we took the un-mapped and aligned against the small RNA reference sequence. The final un-mapped were the reads of interest.
# rRNA filter
bowtie2 --seed=123 --threads=32 -x $rRNA_ref -U $fq -S ${sample_name}_R1.rRNA.sam \
--un-gz=${sample_name}_R1_rFiltered.fq.gz \
> ${sample_name}_R1_rRNA.bowtie2.log 2> ${sample_name}_R1_rRNA.bowtie2.log2
# tRNA and snoRNA filter
bowtie2 --seed=123 --threads=32 -x $tsoRNA_ref \
-U ${sample_name}_R1_rFiltered.fq.gz \
-S ${sample_name}_R1.tsoRNA.sam \
--un-gz=${sample_name}_R1_rtsFiltered.fq.gz \
> ${sample_name}_R1_tsoRNA.bowtie2.log 2> ${sample_name}_R1_tsoRNA.bowtie2.log22.3 Alignment
We took the final un-mapped reads from previous step and aligned against GRCh38.p13 by STAR:
STAR --runThreadN 4 --outSAMattributes NH HI NM MD AS nM jM jI XS \
--genomeDir $star_index \
--readFilesIn $filtered_dir/${sample_name}_R1_rtsFiltered.fq.gz \
--outFileNamePrefix $bam_dir/${sample_name}_ \
--sjdbOverhang 25 \
--outFilterScoreMinOverLread 0 \
--outFilterMatchNminOverLread 0 \
--outFilterMatchNmin 20 \
--readFilesCommand zcat \
--outSAMtype BAM SortedByCoordinate
samtools index $bam_dir/${sample_name}_Aligned.sortedByCoord.out.bamSee next chapter for Ribo-seq specific quality control.