Quick Start for gimap
For more background on gimap and the calculations done here, read here
Requirements
Besides installing the gimap package, you will also need to install
wget if you do not already have it installed. This will allow you to
download the annotation files needed to run gimap.
To install the gimap package you will need to run:
install.packages("gimap")Or you can install the development version from GitHub:
install.packages("remotes")
remotes::install_github("FredHutch/gimap")Loading needed packages
#>
#> Attaching package: 'dplyr'
#> The following objects are masked from 'package:stats':
#>
#> filter, lag
#> The following objects are masked from 'package:base':
#>
#> intersect, setdiff, setequal, unionSet Up
First we can create a folder we will save files to.
output_dir <- "output_timepoints"
dir.create(output_dir, showWarnings = FALSE)
example_data <- get_example_data("count")Setting up data
We’re going to set up three datasets that we will provide to the
set_up() function to create a gimap dataset
object.
-
counts- the counts generated from pgPEN -
pg_ids- the IDs that correspond to the rows of the counts and specify the construct -
sample_metadata- metadata that describes the columns of the counts including their timepoints
counts <- example_data %>%
select(c("Day00_RepA", "Day22_RepA", "Day22_RepB", "Day22_RepC")) %>%
as.matrix()pg_id are just the unique IDs listed in the same
order/sorted the same way as the count data.
Sample metadata is the information that describes the samples and is sorted the same order as the columns in the count data.
sample_metadata <- data.frame(
col_names = c("Day00_RepA", "Day22_RepA", "Day22_RepB", "Day22_RepC"),
day = as.numeric(c("0", "22", "22", "22")),
rep = as.factor(c("RepA", "RepA", "RepB", "RepC"))
)We’ll need to provide example_counts,
pg_ids and sample_metadata to
setup_data().
gimap_dataset <- setup_data(
counts = counts,
pg_ids = pg_ids,
sample_metadata = sample_metadata
)It’s ideal to run quality checks first. The run_qc()
function will create a report we can look at to assess this.
run_qc(gimap_dataset,
output_file = file.path(output_dir, "example_qc_report.Rmd"),
overwrite = TRUE,
quiet = TRUE
)You can take a look at an example QC report here.
gimap_dataset <- gimap_dataset %>%
gimap_filter() %>%
gimap_annotate(cell_line = "HELA") %>%
gimap_normalize(
timepoints = "day"
) %>%
calc_gi()Example output
Genetic interaction is calculated by:
-
pgRNA_target- what gene(s) were targeted by this the original pgRNAs for these data -
mean_expected_cs- the average expected genetic interaction score -
mean_gi_score- the average observer genetic interaction score -
target_type- describes whether the CRISPR design is targeting two genes (“gene_gene”), or a gene and a non targeting control (“gene_ctrl”) or a targeting control and a gene (“ctrl_gene”). -
p_val- p values from the testing whether a double knockout construct is significantly different in its genetic interaction score from single targets.
-
fdr- False discovery rate corrected p values
Plot the results
You can remove any samples from these plots by altering the
reps_to_drop argument.
plot_exp_v_obs_scatter(gimap_dataset)
# Save it to a file
ggsave(file.path(output_dir, "exp_v_obs_scatter.png"))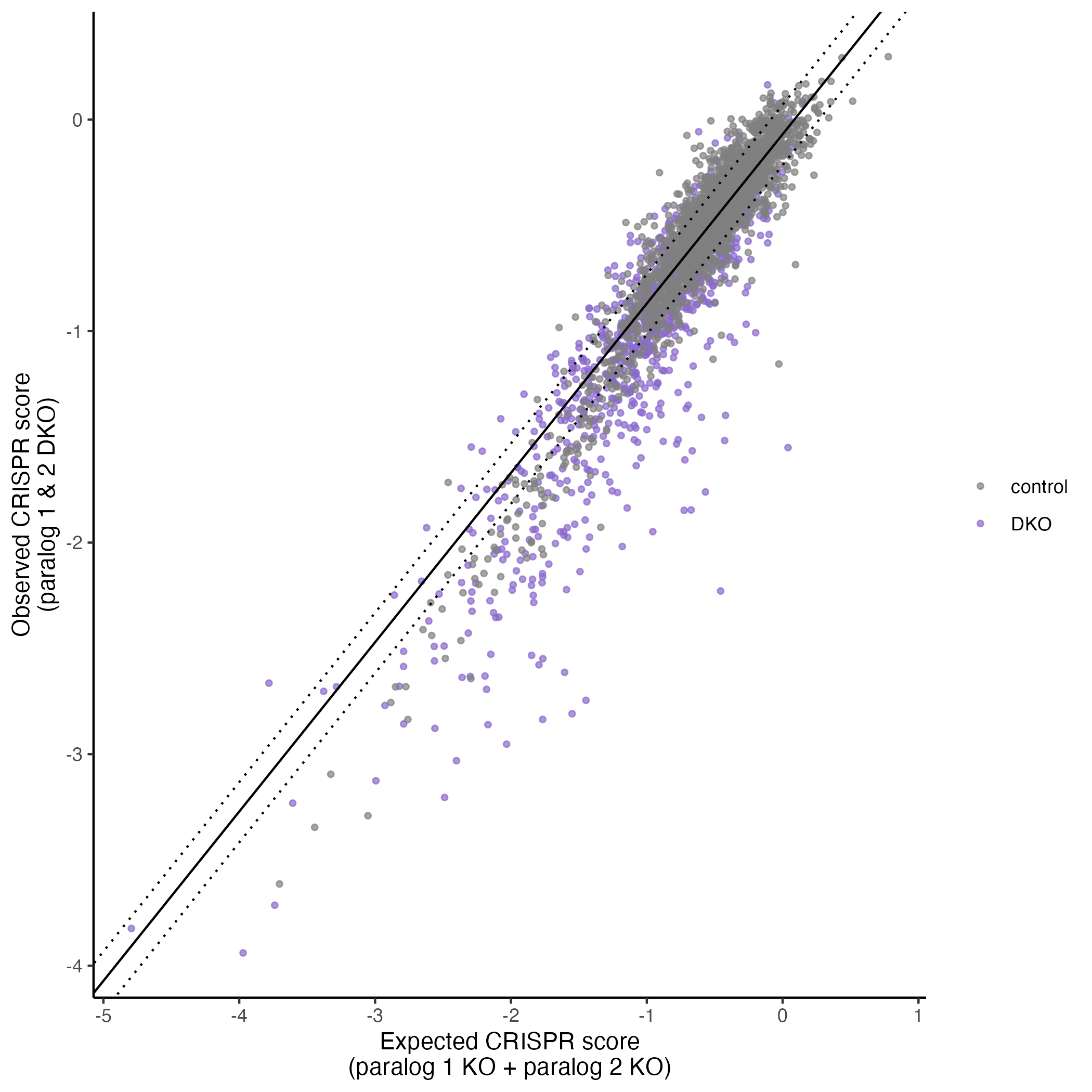
plot_rank_scatter(gimap_dataset)
# Save it to a file
ggsave(file.path(output_dir, "plot_rank_scatter.png"))![]()
plot_volcano(gimap_dataset)
# Save it to a file
ggsave(file.path(output_dir, "volcano_plot.png"))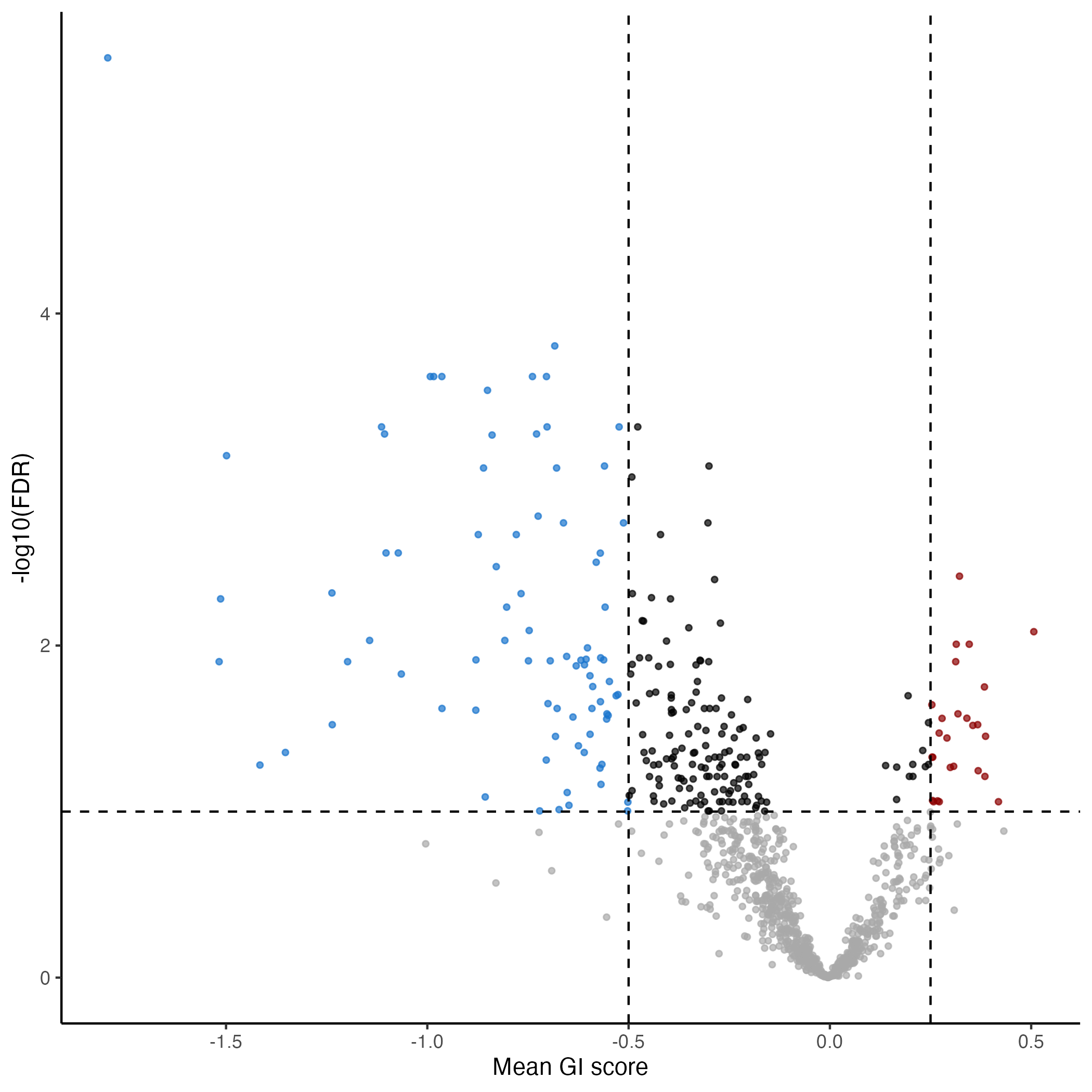
Plot specific target pair
We can pick out a specific pair to plot.
# "CNOT8_CNOT7" is top result so let's plot that
plot_targets(gimap_dataset, target1 = "CNOT8", target2 = "CNOT7")
# Save it to a file
ggsave(file.path(output_dir, "CNOT8_CNOT7.png"))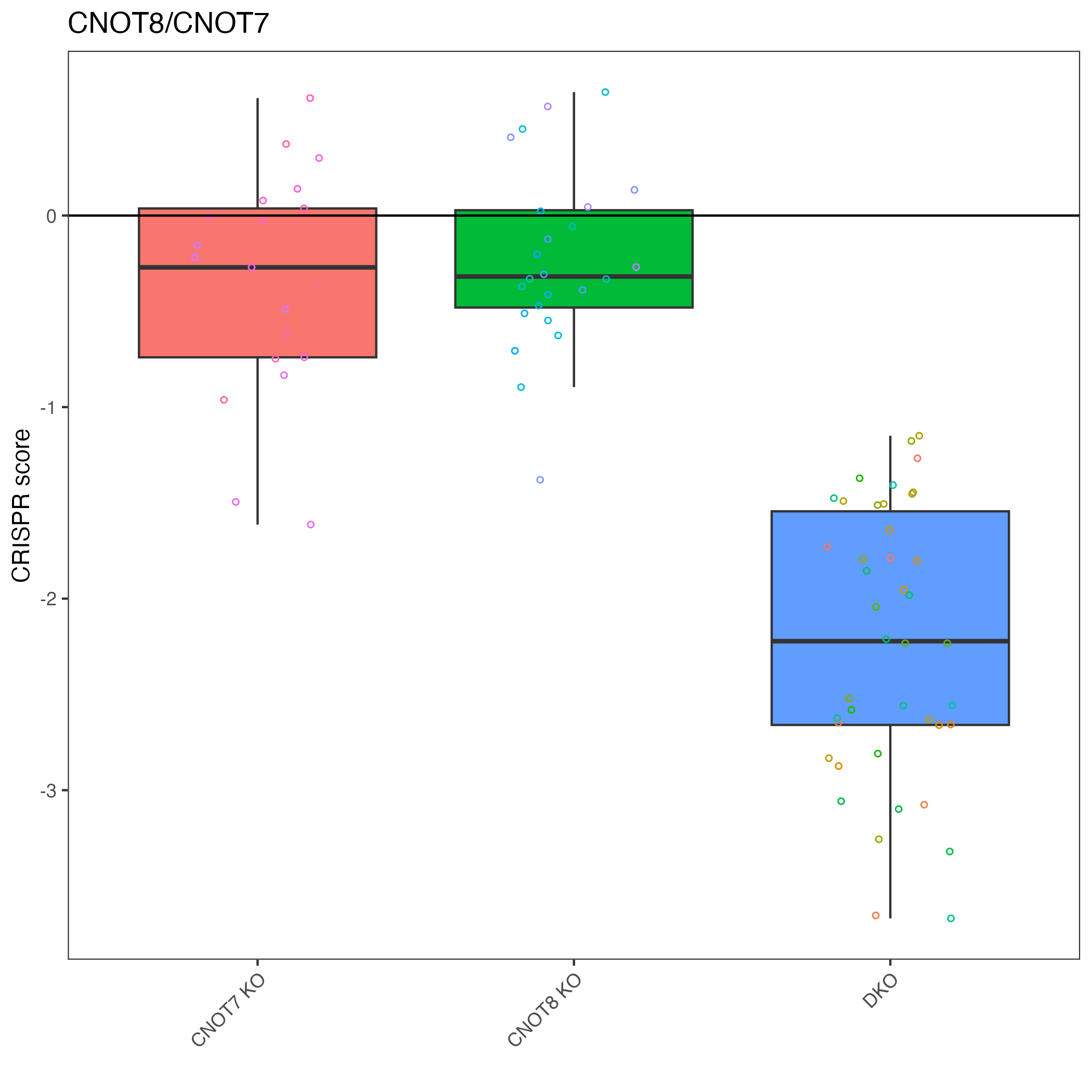
Saving data to a file
We can save all these data as an RDS or the genetic interaction scores themselves to a tsv file.
saveRDS(gimap_dataset, "gimap_dataset_final.RDS")readr::write_tsv(gimap_dataset$gi_scores, "gi_scores.tsv")Session Info
This is just for provenance purposes.
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: x86_64-apple-darwin20
#> Running under: macOS 15.3.2
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-x86_64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: America/New_York
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] dplyr_1.1.4 gimap_1.0.3
#>
#> loaded via a namespace (and not attached):
#> [1] gtable_0.3.6 jsonlite_1.8.9 compiler_4.4.0 tidyselect_1.2.1
#> [5] stringr_1.5.1 snakecase_0.11.1 tidyr_1.3.1 jquerylib_0.1.4
#> [9] systemfonts_1.2.1 scales_1.3.0 textshaping_1.0.0 yaml_2.3.10
#> [13] fastmap_1.2.0 ggplot2_3.5.1 R6_2.5.1 generics_0.1.3
#> [17] knitr_1.49 htmlwidgets_1.6.4 tibble_3.2.1 janitor_2.2.1
#> [21] desc_1.4.3 openssl_2.3.2 munsell_0.5.1 lubridate_1.9.4
#> [25] RColorBrewer_1.1-3 bslib_0.8.0.9000 pillar_1.10.1 rlang_1.1.5
#> [29] stringi_1.8.4 cachem_1.1.0 xfun_0.50 fs_1.6.5
#> [33] sass_0.4.9 timechange_0.3.0 cli_3.6.3 pkgdown_2.1.1
#> [37] magrittr_2.0.3 digest_0.6.37 grid_4.4.0 rstudioapi_0.17.1
#> [41] askpass_1.2.1 lifecycle_1.0.4 vctrs_0.6.5 pheatmap_1.0.12
#> [45] evaluate_1.0.3 glue_1.8.0 ragg_1.3.3 colorspace_2.1-1
#> [49] purrr_1.0.4 rmarkdown_2.29 httr_1.4.7 tools_4.4.0
#> [53] pkgconfig_2.0.3 htmltools_0.5.8.1